(本文中国大陆首发于 2026 年 5 月 28 日 BY 润联消毒研究院)
依据 EU GMP Annex 1(2022年修订版)及 US FDA 21 CFR Part 211
2026年5月

一、引言:对高效杀芽孢去污技术的迫切需求
无菌药品的无菌生产——涵盖注射用生物制剂、眼用溶液及冻干制剂——要求生产环境中的微生物污染被降至绝对最低水平。与应用于最终产品的终端灭菌工艺不同,无菌工艺的核心在于生产环境本身的无菌状态,因此污染控制策略(Contamination Control Strategy,CCS)是产品质量与患者安全的基石。
在现有的去污技术中,气化过氧化氢(VHP)凭借其卓越的杀芽孢效力、良好的材料兼容性,以及分解产物仅为水蒸气和氧气、无有害残留的独特优势,已取得无可比拟的领先地位。VHP能够渗透至隔离器(Isolator)、限制进出屏障系统(RABS)及A/B级洁净室内的复杂几何空间,使其在现代无菌生产中不可或缺。
具有里程碑意义的法规转折点出现于2022年8月欧盟GMP附录1修订版的发布。该修订版史无前例地强调了整体性CCS理念,并在屏障技术的框架下明确提及了VHP类去污剂。与此同时,美国FDA在21 CFR Part 211及相关指南文件中持续对无菌生产的环境控制提出严格要求。这两大法规体系共同构成了VHP程序设计、验证与维护的监管框架。
二、法规支持与法律框架
2.1 欧盟GMP附录1(2022年修订版)
欧盟委员会于2022年8月22日正式发布GMP附录1《无菌药品生产》修订版全文,并于2023年8月25日正式生效。此次修订是二十余年来对无菌生产法规最为全面的一次更新,也是欧洲乃至全球范围内推动VHP技术应用的首要法规驱动力。
第2节将“污染控制策略(CCS)”作为统一性法规概念正式引入,其原文如下:
"A contamination control strategy (CCS) should be implemented across the facility to define all critical control points and assess the effectiveness of all the controls (design, procedural, technical and organisational) and monitoring measures employed to manage risks to the sterility of the product... The CCS should identify and implement the appropriate controls to assure product quality; these may include: [...] use of appropriate technologies e.g. Rapid Microbial Methods (RMM), environmental monitoring, blow-fill-seal, barrier and isolator technologies."
— EU GMP Annex 1 (2022), Section 2.1
VHP的主要应用场景——隔离器与RABS——在附录1第8节(隔离器与密闭RABS)中有明确规定:
"The transfer of materials into and out of the unit is one of the greatest potential sources of contamination. In general, the area inside the isolator is the place where operations are carried out. Entry of materials into the isolator should only be performed through transfer devices. The isolator and its transfer system should be validated. Decontamination methods should be validated. Particular attention should be paid to validate both the materials used and the contact surfaces..."
— EU GMP Annex 1 (2022), Section 8.7
此外,附录1在洁净室去污情境下直接涉及生物污染与杀芽孢剂的相关要求:
"Disinfectants and detergents used in Grade A and B areas should be sterile prior to use. Disinfectants and detergents used in Grade C and D areas should be of appropriate quality. Where required by the CCS, sporicidal agents should also be used."
— EU GMP Annex 1 (2022), Section 4.36
上述条款明确要求,当风险评估有此需要时,须在A/B级区域使用杀芽孢剂——而VHP正是目前经过最广泛验证和认可的杀芽孢剂——这实际上将VHP或同等技术确立为最高级别洁净室的法规预期。
2.2 美国FDA法规框架
在美国,无菌生产主要受21 CFR Part 211(《成品药现行药品生产质量管理规范》)以及FDA的里程碑性指南文件——《行业指南:通过无菌工艺生产的无菌药品——现行药品生产质量管理规范》(2004年9月,简称“无菌工艺指南”)的约束。
21 CFR § 211.42(c) 确立了环境控制的基本法律要求,其原文如下:
"Operations shall be performed within specifically defined areas of adequate size. There shall be separate or defined areas or such other control systems for the firm's operations as are necessary to prevent contamination or mixups during the course of the following procedures: [...] (10) Aseptic processing, which includes as appropriate: (i) Floors, walls, and ceilings of smooth, hard surfaces that are easily cleanable; (ii) Temperature and humidity controls; (iii) An air supply filtered through high-efficiency particulate air filters under positive pressure, regardless of whether flow is laminar or nonlaminar; (iv) A system for monitoring environmental conditions; (v) A system for cleaning and disinfecting the room and equipment to produce aseptic conditions."
— 21 CFR § 211.42(c)(10)
FDA《无菌工艺指南》(2004年)进一步阐述了杀芽孢剂在洁净室中的应用:
"Use of a sporicidal agent should be a component of the program for any aseptic processing room... Bacterial endospores are extremely resistant to disinfectants and are used as challenge agents to validate sterilization and decontamination processes."
— FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing (2004), Section X.B
FDA的工艺验证指南(2011年)进一步强化了生命周期管理方法,对VHP循环开发具有直接指导意义:
"Process validation is the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product... Each manufacturer should judge whether it has collected sufficient evidence to support a conclusion that its manufacturing process is adequately controlled."
— FDA Guidance for Industry: Process Validation: General Principles and Practices (2011), p. 5
2.3 协调标准:ISO与USP
除GMP法规外,ISO 14937:2009(《医疗器械灭菌——灭菌剂特性及医疗器械灭菌过程开发、验证和常规控制的通用要求》)和ISO 22441(专门针对VHP在医疗器械中的应用)为VHP验证提供了技术框架。USP <1229.12>(《药典品种的灭菌——新型灭菌方法》)亦涵盖了包括过氧化氢蒸气在内的气态和蒸气相灭菌剂的适用原则。
PDA技术报告第51号(《气体和蒸气相去污过程用生物指示剂:意义、选择与使用》)被普遍引用于VHP验证方案中,就生物指示剂(BI)的选择、放置与结果判读提供了权威指导。
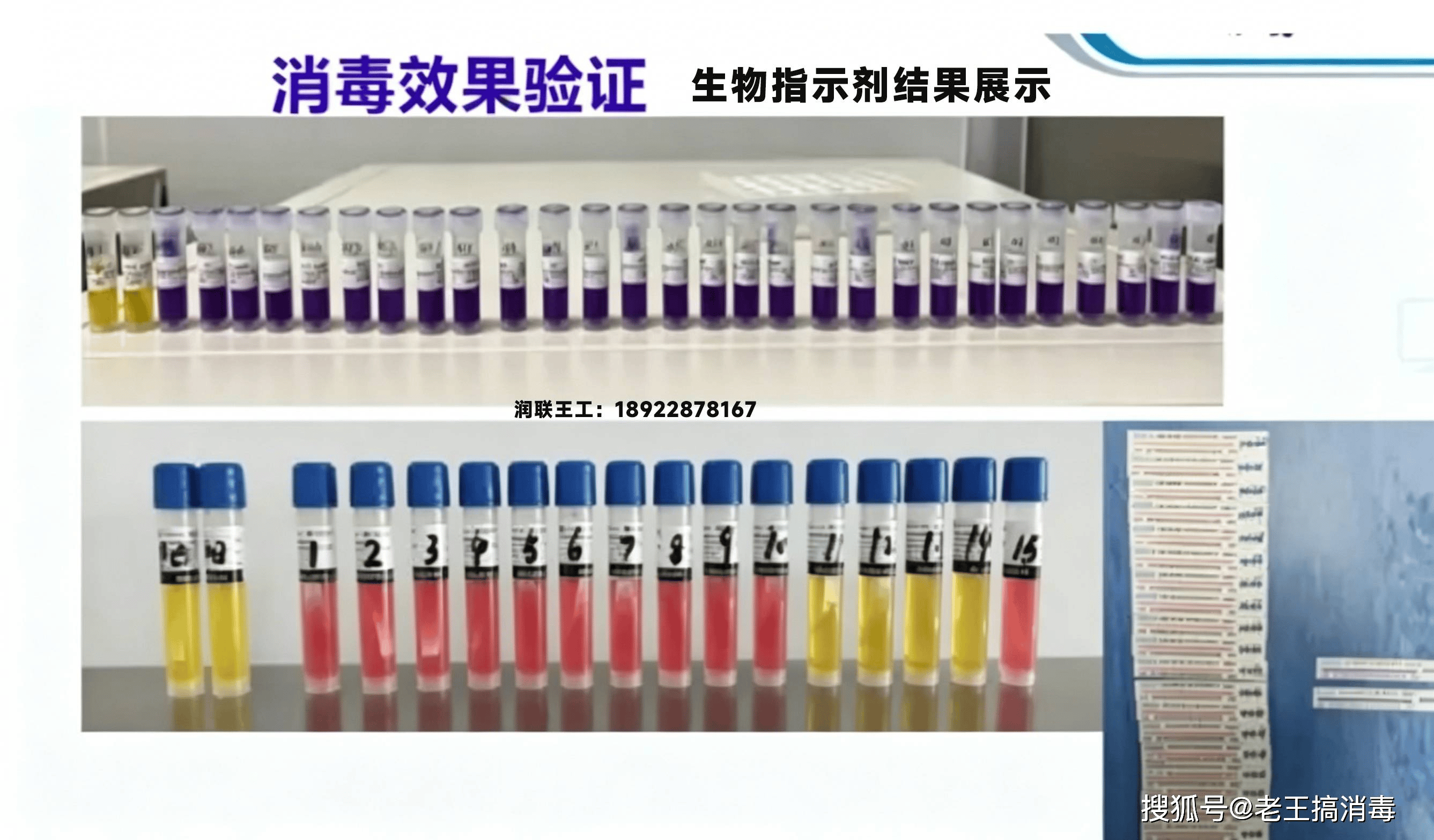 生物指示剂
生物指示剂
三、具体法规要求
3.1 适用范围:VHP的强制使用场景
对VHP使用的法规预期集中于发生无菌产品或组件无菌操作的环境。以下环境具有最强的VHP验证法规压力:
- A级隔离器:用于无菌灌装、塞子插入及容器密封的主要封闭装置。附录1第8节将隔离器去污作为设计的基本要求。
- A/B级RABS:开放式和密闭式RABS均需要有文件记录的杀芽孢去污程序,并验证达到适当的对数降低值。
- B级洁净室:作为A级操作的背景环境,需定期进行杀芽孢处理,特别是在维护作业、环境偏差或长时间停工后,这是CCS的组成部分。
- 传递舱与气闸:进入A/B级环境的所有物料和组件传递系统均需进行去污验证。
- 冻干机腔体:在无菌条件下将产品装入和卸出冻干机是关键控制点,须有经过验证的去污方案。
3.2 验证要求
3.2.1 微生物致死标准
法规机构与行业标准均指向同一最低杀芽孢效力标准:对嗜热脂肪地芽孢杆菌(Geobacillus stearothermophilus)或枯草芽孢杆菌(Bacillus atrophaeus)芽孢的最低6对数(6-log₁₀)降低。EU附录1第8.8节明确规定:
"The effectiveness of the decontamination cycle used should be validated, and the validation process should include exposure of biological indicators (BIs) at defined worst-case positions within the enclosure... The validation should demonstrate that the required log reduction is achieved consistently."
— EU GMP Annex 1 (2022), Section 8.8
FDA《无菌工艺指南》通过其关于灭菌原则的论述及对文件化致死数据的要求,隐含地支持了这一标准。PDA技术报告第51号明确建议关键区域去污应达到最低6对数降低。
3.2.2 生物指示剂要求
嗜热脂肪地芽孢杆菌(ATCC 7953)芽孢是VHP工艺公认的生物指示剂,因其在VHP过程中表现出经充分验证的抗性特征。主要法规与标准要求包括:
- BI菌落数:每载体≥10⁶ CFU(须经制造商分析证书核实)
- D值:须在特定VHP工艺条件(浓度、温度、湿度)下进行表征
- BI放置位置:须由最差情况分布研究确定,包括循环开发阶段的数据
- 阳性对照:每次验证运行至少设置一个阳性对照,以确认BI的活性
- 可追溯性:每批BI须可追溯至制造商证书及有效期
附录1同时要求BI放置策略须通过分布研究加以证明:
"The number and positioning of the biological indicators should be justified based on the worst-case locations identified for the decontamination agent."— EU GMP Annex 1 (2022), Section 8.8
3.2.3 化学指示剂与浓度监测
除BI外,验证方案还须包含化学浓度监测。法规指南要求使用经验证的传感器技术(电化学或红外传感器)对整个工艺循环中H₂O₂蒸气浓度进行实时测量。经验证的浓度范围须被明确定义,并在验证运行中被证明具有可重复性。
3.3 循环开发与表征要求
EU GMP附录1与ISO 14937共同要求在验证前开展系统化的循环开发程序,具体包括:
- 四个循环阶段的定义:
第一阶段 — 调节(Conditioning):湿度与温度稳定化
第二阶段 — 去污(Decontamination):H₂O₂注入与暴露
第三阶段 — 驻留(Dwell):维持H₂O₂浓度
第四阶段 — 通风(Aeration):将H₂O₂去除至安全残留水平
- H₂O₂残留限值:须满足职业暴露限值(OEL)1 ppm(OSHA/ACGIH标准)或产品接触表面残留限值,以更严格者为准。
- 分布研究:对整个封闭空间内H₂O₂浓度分布进行图谱化,以识别最差情况低浓度区域。
- 重现性运行:至少三次连续成功的验证运行(或经风险评估另行论证)。
四、实施注意事项与最佳实践
4.1 实施前准备:风险评估与CCS整合
VHP的成功实施始于基于风险的方法,并将其整合至工厂整体CCS中。PIC/S和EU GMP均强调,技术选择必须以风险评估为驱动。关键的实施前活动包括:
- 工厂风险评估:对所有潜在污染途径进行系统图谱化
- 材料兼容性评估:对所有表面、弹性体、电子元件和产品接触材料开展评估(H₂O₂是强氧化剂,可能随时间降解某些材料)
- HVAC整合审查:VHP循环要求HVAC系统被隔离或专门配置,以防止H₂O₂渗入非目标区域
- 安全评估:H₂O₂蒸气在1 ppm以上被列为危险气体;安全程序和联锁装置须在调试前明确规定
- 法规申报策略:在某些司法管辖区,去污方法的变更可能需要在上市许可中事先获批或通知
4.2 设备选型与确认
VHP发生器的选型须满足特定应用需求。关键技术参数如下表所示:
 关键技术参数
关键技术参数
4.3 验证方案执行
验证方案须在执行前获得审批(EU GMP第4章;21 CFR § 211.68)。关键执行注意事项包括:
- IQ/OQ/PQ结构:安装确认(IQ)须核实发生器规格是否符合采购订单;运行确认(OQ)须在规定条件下确立循环参数;性能确认(PQ)须证明在最差条件下实现BI 6-log降低。
- 温湿度控制:VHP效力对相对湿度(RH)高度敏感。最优RH通常为30–60%;低于30% RH会显著降低效力。循环前预调节至目标RH是关键步骤,须经过验证和控制。
- 最差情况位置:须包括气流速度最低处、距VHP注射点最远处,以及几何遮挡最复杂的区域(如搁板支架下方、过滤器外壳内部)。
- 文件记录:所有原始数据,包括传感器轨迹、BI结果、环境条件和操作人员信息,须同步记录并按21 CFR Part 11/EU附录11归档保存。
4.4 日常运行与定期再验证
经过验证的VHP程序需要对验证状态进行持续维护。法规要求包括:
- 定期再验证:由变更控制事件触发(设备更换、工厂改造、循环参数变更)以及基于风险的定期计划(关键应用通常为每年一次)。
- 日常循环监测:每个生产循环须生成循环报告,包含H₂O₂浓度与时间曲线、温度、湿度和通风终点数据,与经验证的验收标准进行比对。
- 环境监测趋势分析:VHP后的环境监测结果须作为污染控制体系的组成部分进行趋势分析和审查,以确认循环的持续有效性。
- 材料兼容性再评估:达到显著累计H₂O₂暴露量后(通过循环计数器追踪),须对材料兼容性进行重新评估并记录。
- 人员培训:所有执行VHP循环的操作人员须经相关SOP培训并具备资质;培训记录须按21 CFR § 211.68及EU GMP第2章保存。
4.5 常见问题与风险缓解
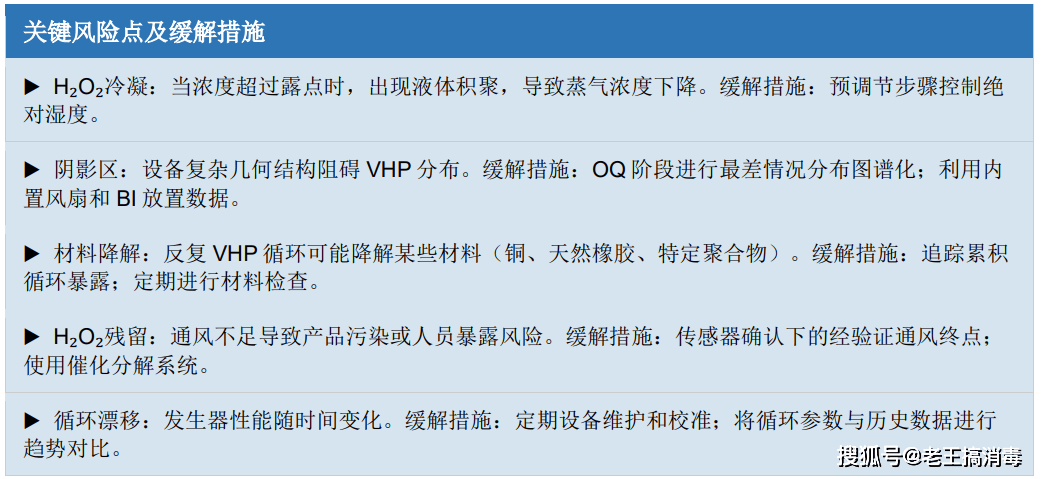 常见问题与风险缓解
常见问题与风险缓解
五、市场最新技术与创新进展
5.1 新一代VHP发生器平台
商业VHP发生器市场在过去五年中经历了显著的技术进步。来自全球各地的公司,已引入直接应对GMP合规挑战的新功能:
- 闭环浓度控制(Closed-loop concentration control):集成H₂O₂传感器与注射速率调节之间的实时反馈,在整个驻留阶段将浓度维持在设定的浓度窗口内,显著提升循环重现性并降低验证变异性。来自中国的某品牌SVHP已经可以实现通过浓度监控,控制H₂O₂发生量的功能。
- 符合21 CFR Part 11/EU Annex 11的集成数据管理:具有密码学安全审计追踪的电子批记录,消除手动数据转录风险。
- 小型化便携式发生器平台:设计用于在洁净室环境内使用而无需穿透HVAC系统的紧凑型机组(如STERIS VHP® 1000ED,欧菲姆®SVHP-M),将应用场景扩展至临时隔离和移动使用场景。
5.2 先进传感器与监测技术
传感器技术是VHP验证和日常监测领域近年来影响最大的创新领域之一:
- 电化学H₂O₂传感器:与红外传感器相比,提供更优越的长期稳定性和更低的灵敏度漂移,为趋势分析提供更可靠的逐循环数据。德尔格(过氧化氢传感器系列),欧菲姆(FDH01)等公司的产品以高灵敏度及可科学校准的方法提供实时ppm级精度。
- 分布式无线传感器阵列:使用无线H₂O₂集成系统(如Mesa Labs SporView®及 欧菲姆®BioCenter)进行多点在线实时监测,将传统的单一位置浓度表象弊端彻底解决,能够为使用者提供整个洁净区的H₂O₂分布浓度实时在线监测。
- 产品接触表面H₂O₂残留监测:新型比色法和电化学检测系统用于表面残留核实,在通风终点传感器数据之外,进一步提供直接的表面污染确认。
5.3 人工智能与机器学习的集成应用
人工智能与数据分析技术正在开始变革VHP循环管理模式,特别是在生命周期验证的背景下:
- 预测性循环优化(Predictive cycle optimization):机器学习算法基于历史循环数据(浓度曲线、BI结果、环境条件)进行训练,以预测最优注射参数——在确保稳定6-log效力的同时最小化通风时间,直接支持FDA的工艺验证生命周期方法。
- 异常检测与预测性维护(Anomaly detection and predictive maintenance):基于AI的发生器性能指标监测,在循环失败前检测早期部件退化(注射系统污堵、传感器漂移),支持持续工艺确认(CPV)程序。
- 数字孪生仿真(Digital twin simulation):基于计算流体动力学(CFD)的隔离器几何和气流模式数字孪生模型,用于在物理验证运行前通过计算机模拟预测H₂O₂分布并识别最差情况区域,降低材料和时间成本。
5.4 新型配方与应用创新
除硬件创新外,配方与应用方法的创新也在拓展VHP的应用边界:
- 小于8%的过氧化氢液体:长期以来,VHP的使用受制于各国对危险化学品管控,但是限于现有蒸发技术瓶颈,其损耗较大,因此35%的过氧化氢溶液是使用首选。随着新型蒸发技术的问世,符合非危化品(即H₂O₂浓度小于8%)的过氧化氢溶液使用于VHP已经出现并开始推广。(如:NOVUSWATER®NOCOCIDE及Bosch专用H₂O₂溶液)需要注意的是,这些溶液通常需要配合厂商允许使用目录中的设备厂家产品。
- 连续生产环境中的VHP应用:开发与连续无菌生产(CAM)工艺设计兼容的VHP去污方案,这是FDA新兴技术项目重点支持的快速增长领域。
- 先进治疗药品(ATMP)领域的VHP应用:为细胞和基因治疗生产中的洁净室去污开发专用低温VHP循环,以规避传统高温或辐射方法与环境中生物材料不兼容的问题。
六、法规检查预期
基于已公开的FDA警告信(Warning Letters)、EMA检查观察项及MHRA检查结论,法规检查员在现场检查中持续审查VHP程序的以下方面:
- 验证范围不足:未将所有物料传递系统、传递舱和气闸纳入经验证的去污程序。检查员曾因制造商仅对主隔离器腔体进行验证而对传递系统未作验证而提出缺陷。
- 最差情况BI放置论证不充分:方案缺乏基于分布图谱数据的BI位置选择的书面科学依据。
- 缺失21 CFR Part 11合规性:电子循环记录缺少审计追踪、用户访问控制,或可被未授权修改。
- 变更后未启动再验证:隔离器配置、HVAC参数或H₂O₂发生器设备发生变更后未触发基于变更控制的再验证。
- 去污后环境监测不充分:缺少经文件记录的、可趋势分析的环境监测程序,无法确认日常生产中VHP循环的持续有效性。
FDA近期与无菌工艺失败相关的警告信(2022–2024年)持续将“污染控制策略不充分”及“杀芽孢去污工艺验证不足”列为主要缺陷项。VHP程序的设计与记录因此必须能够经受最高级别法规审查的检验。
七、结论
蒸汽化过氧化氢(VHP)已在无菌制药生产领域确立了其杀芽孢去污技术的主导地位,并获得以EU GMP Annex 1(2022年)和美国FDA 21 CFR Part 211无菌工艺要求为基础的完善且持续演进的法规框架支撑。法规要求清晰且严格:VHP程序必须经过充分验证、严格记录,并被整合入全面的污染控制策略中。
成功实施VHP需要高度关注材料兼容性、循环开发规范性、最差情况BI放置的科学性以及符合21 CFR Part 11要求的数据管理。随着技术的持续演进——包括AI赋能的循环优化、无线实时BI监测和基于CFD的数字孪生仿真——制药生产商拥有了日益丰富的工具包,可以以前所未有的信心实现并维持经验证的VHP性能。
日趋严格的法规预期与快速进步的技术能力相互融合,使VHP不仅仅是一项合规要求,更成为企业的战略质量资产——使生产商能够以超越任何以往去污方法的确定性,守护产品无菌性、患者安全与生产运营的持续性。
 欧菲姆气化过氧化氢消毒设备
欧菲姆气化过氧化氢消毒设备
参考文献及法规文件
1. European Commission. (2022). EudraLex Volume 4, GMP Annex 1: Manufacture of Sterile Medicinal Products. European Commission, Brussels. Effective August 25, 2023.
2. U.S. Food and Drug Administration. (2004). Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice. FDA, Silver Spring, MD.
3. U.S. Food and Drug Administration. (2011). Guidance for Industry: Process Validation: General Principles and Practices. FDA, Silver Spring, MD.
4. Code of Federal Regulations. 21 CFR Part 211 — Current Good Manufacturing Practice for Finished Pharmaceuticals. U.S. Government Publishing Office.
5. International Organization for Standardization. (2009). ISO 14937: Sterilization of Health Care Products — General Requirements for Characterization of a Sterilizing Agent. ISO, Geneva.
6. Parenteral Drug Association. Technical Report No. 51: Biological Indicators for Gas and Vapor-Phase Decontamination Processes. PDA, Bethesda, MD.
7. United States Pharmacopeia. <1229.12> Sterilization of Compendial Articles — New Sterilization Methods. USP-NF.
8. Pharmaceutical Inspection Co-operation Scheme (PIC/S). PI-007-6: Recommendations on Sterility Testing. PIC/S Secretariat, Geneva.
9. Vaisala Corporation. (2023). Hydrogen Peroxide Measurement for Biodecontamination Applications. Application Note.
10. STERIS Corporation. (2024). VHP Technology Platform Technical Documentation. STERIS Life Sciences.
11. European Medicines Agency. (2023). Overview of Comments Received on Draft Annex 1 to the EU GMP Guidelines. EMA, Amsterdam.
12. FDA Warning Letters Database (2022–2024). Aseptic Processing Deficiencies. Accessed via FDA.gov.